"All we have discovered is that it starts with a single individual - always a child - and then spreads explosively, like the formation of crystals round the first nucleus in a saturated solution." Arthur C. Clarke (Childhood's End, 1953)
In The News
09/07/2016 |
5 publications have been added:Nickel Complexes of Pyridine-Functionalized N-Heterocyclic Carbenes: Syntheses, Structures, and Activity in Electrocatalytic Hydrogen Production Eur. J. Inorg. Chem. (2016), , Ahead of Print - link Changes in the luminescence emission of α,β-unsaturated acrylonitrile derivatives: morphology, polymorphism and solvent effect CrystEngComm (2016), , Advance Article - link Localized Mixed-Valence and Redox Activity within a Triazole-Bridged Dinucleating Ligand upon Coordination to Palladium Chem. Eur. J. (2016), , Ahead of Print - link Reversible Redox Chemistry and Catalytic C(sp3)–H Amination Reactivity of a Paramagnetic Pd Complex Bearing a Redox-Active o-Aminophenol-Derived NNO Pincer Ligand Inorg. Chem. (2016), 55, 8603-8611 - link The Influence of Peripheral Substituent Modification on PV, MnIII, and MnV(O) Corrolazines: X-ray Crystallography, Electrochemical and Spectroscopic Properties, and HAT and OAT Reactivities Inorg. Chem. (2016), 55, 8646-8660 - link |
08/15/2016 |
2 publications have been added:A C–F Bond Directed Diels–Alder Reaction J. Org. Chem. (2016), , Article ASAP - link Arene C(sp2)-H Metalation at NiII Modeled with a Reactive PONCPh Ligand Inorg. Chem. Int. Ed. (2016), 55, 8041-8047 - link |
07/26/2016 |
2 publications have been added:Well-Defined Dinuclear Gold Complexes for Preorganization-Induced Selective Dual Gold Catalysis Angew. Chem. Int. Ed. (2016), , Ahead of Print - link Isocyanide or nitrosyl complexation to hemes with varying tethered axial base ligand donors: synthesis and characterization J. Biol. Inorg. Chem. (2016), , Ahead of Print - link |
07/19/2016 |
1 publication has been added:The close interaction of a C-F bond with a carbonyl π–system: Attractive, repulsive, or both? J. Fluorine Chem. (2016), 188, 126-130 - link |
07/11/2016 |
1 publication has been added:Chemical Swarming: Depending on Concentration, an Amphiphilic Ruthenium Polypyridyl Complex Induces Cell Death via Two Different Mechanisms Chem. Eur. J. (2016), , Early View - link |
07/05/2016 |
1 publication has been added:Highly regiocontrolled and stereocontrolled syntheses of polysubstituted aminocyclohexanes: mild inverse-electron-demand Diels–Alder cycloadditions of electrophilic 2-pyridones Tetrahedron Lett. (2016), , Ahead of Print - link |
06/27/2016 |
1 publication has been added:Redox-Active-Ligand-Mediated Formation of an Acyclic Trinuclear Ruthenium Complex with Bridging Nitrido Ligands Angew. Chem. Int. Ed. (2016), , Ahead of Print - link |
06/16/2016 |
3 publications have been added:A Distance Dependence to Lateral Self-Exchange Across Nanocrystalline TiO2. A Comparative Study of Three Homologous RuIII/II Polypyridyl Compounds J. Phys. Chem. C,. (2016) - link Aromaticity Competition in Differentially Fused Borepin-Containing Polycyclic Aromatics J. Org. Chem. (2016) - link RPeroxo and Superoxo Moieties Bound to Copper Ion: Electron-Transfer Equilibrium with a Small Reorganization Energy J. Am. Chem. Soc. (2016) - link |
05/23/2016 |
2 publications have been added:Ratiometric Thermometer Based on a Lanthanoid Coordination Polymer Eur. J. Inorg. Chem. (2016), , Early View - link Positioning a Carbon–Fluorine Bond over the π Cloud of an Aromatic Ring: A Different Type of Arene Activation Angew. Chem. Int. Ed. (2016) - link |
03/15/2016 |
1 publication has been added:Photocatalytic Oxygenation of Substrates by Dioxygen with Protonated Manganese(III) Corrolazine Inorg. Chem. (2016) - link |
03/14/2016 |
5 publications have been added:Copper(I) Complexes of Naphthyl-Substituted Fluorinated Trispyrazolylborate Ligands with Ethene and Carbon Monoxide Eur. J. Inorg. Chem. (2016) link Dioxygen Activation by a Macrocyclic Copper Complex Leads to a Cu2O2 Core with Unexpected Structure and Reactivity Chem. Eur. J. (2016) - link Photoinitiated Reactivity of a Thiolate-Ligated, Spin-Crossover Nonheme {FeNO}7 Complex with Dioxygen J. Am. Chem. Soc. (2016) - link Innocent BN bond substitution in anthracene derivatives Org. Biomol. Chem. (2016) - link On the Chemistry and Physical Properties of Flux and Floating Zone Grown SmB6 Single Crystals Sci. Rep. (2016) - link |
01/19/2016 |
Congratulations to Danny Broere from the van der Vlugt lab (University of Amsterdam) for his recently published article in Angewandte Chemie International Edition (IF: 11.261) - linkMetal–Metal Interactions in Heterobimetallic Complexes with Dinucleating Redox-Active LigandsD. L. J. Broere, D. K. Modde, E. Blokker, M. A. Siegler, J. I. van der Vlugt Congratulations to Prof. Kerber and his research group from Bucknell University for his recently published article in Inorg. Chem (IF: 4.762) - linkEnhanced Stability of the FeII/MnII State in a Synthetic Model of Heterobimetallic Cofactor AssemblyW.D. Kerber, J. T. Goheen, K. A. Perez, M. A. Siegler |
11/18/2015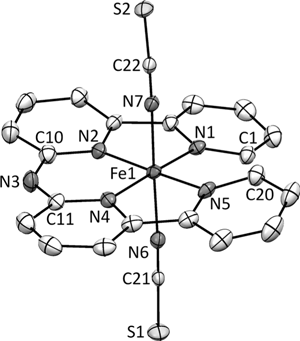 |
Congratulations to Dr. Zheng from the Bonnet lab (Leiden University, The Netherlands) for his recently published article in Chem. Eur. J. (IF: 5.731) - linkStabilization of the Low-Spin State in a Mononuclear Iron(II) Complex and High-Temperature Cooperative Spin Crossover Mediated by Hydrogen BondingS. Zheng, N. R. M. Reintjens, M. A. Siegler, O. Roubeau, E. Bouwman, A. Rudavskyi, R. W. A. Havenith, S Bonnet Abstract: The tetrapyridyl ligand bbpya (bbpya=N,N-bis(2,2′-bipyrid-6-yl)amine) and its mononuclear coordination compound [Fe(bbpya)(NCS)2] (1) were prepared. According to magnetic susceptibility, differential scanning calorimetry fitted to Sorai’s domain model, and powder X-ray diffraction measurements, 1 is low-spin at room temperature, and it exhibits spin crossover (SCO) at an exceptionally high transition temperature of T1/2=418 K. Although the SCO of compound 1 spans a temperature range of more than 150 K, it is characterized by a wide (21 K) and dissymmetric hysteresis cycle, which suggests cooperativity. The crystal structure of the LS phase of compound 1 shows strong N-H⋅⋅⋅S intermolecular H-bonding interactions that explain, at least in part, the cooperative SCO behavior observed for complex 1. DFT and CASPT2 calculations under vacuum demonstrate that the bbpya ligand generates a stronger ligand field around the iron(II) core than its analogue bapbpy (N,N'-di(pyrid-2-yl)-2,2′-bipyridine-6,6′-diamine); this stabilizes the LS state and destabilizes the HS state in 1 compared with [Fe(bapbpy)(NCS)2] (2). Periodic DFT calculations suggest that crystal-packing effects are significant for compound 2, in which they destabilize the HS state by about 1500 cm−1. The much lower transition temperature found for the SCO of 2 compared to 1 appears to be due to the combined effects of the different ligand field strengths and crystal packing. |
11/16/2015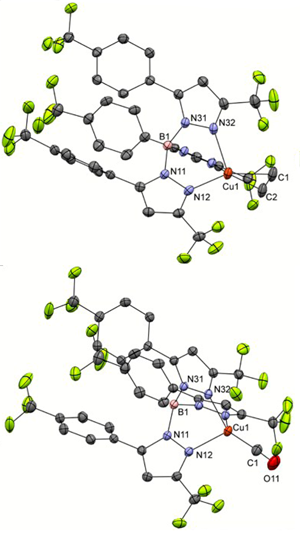 |
Congratulations to Thomas van Dijkman from the Bouwman lab (Leiden University, The Netherlands) for his recently published article in Dalton Trans. (IF: 4.197) - linkHighly tunable fluorinated trispyrazolylborates [HB(3-CF3-5-{4-RPh}pz)3]– (R = NO2, CF3, Cl, F, H, OMe and NMe2) and their copper(I) complexes.T. F. van Dijkman, M. A. Siegler, E. Bouwman Abstract: The ethene and carbon monoxide adducts of copper(I) with seven trispyrazolylborate ligands ([HB(3-CF3-5-{4-RPh}pz)3]–; R = NO2 (4a), CF3 (4b), Cl (4c), F (4d)¸ H (4e)¸ OMe (4f) and NMe2 (4g)) were synthesized and characterized. The ligands were synthesized from their corresponding pyrazoles and sodium tetrahydridoborate and were obtained as solvent adducts of their sodium salts after workup. When the pyrazole with the most electron-withdrawing substituent (R = NO2) is used the asymmetric ligand [HB(3-CF3-5-{4-NO2Ph}pz)2(3-{4-NO2Ph}-5-CF3pz)]– (4a’)is formed as the major product. Copper(I) complexes with ethene or CO as a co-ligand were prepared in good yields and were structurally characterized using 1H NMR, 13C NMR and infrared spectroscopy. Single crystal X-ray crystallography analyses revealed the structures of Na4a’, Na4b, four copper ethene complexes and four copper carbon monoxide complexes. The structures of the copper(I) complexes show CuI ions in pseudo-tetrahedral coordination environments consisting of three nitrogen atoms of the trispyrazolylborate ligand and the carbon monoxide or η2–coordinated ethene ligands, with nearly identical coordination environments around the CuI ion. The compound [Na(4a’)(H2O)]n crystallizes as one-dimensional chains with intermolecular Na•••O2N interactions. The sodium ions were found in severely distorted octahedral geometries with three nitrogen atoms from the trispyrazolylborate ligand, one aqua ligand and two oxygen atoms from the nitro group of an adjacent molecule. The compound [Na2(4b)2(µ-H2O)2] crystallizes as a centrosymmetric water-bridged dimer: two five-coordinate square-pyramidal sodium ions each are coordinated facially by three nitrogen atoms from a trispyrazolylborate ligand and two bridging water ligands. Below the base of the pyramidal structure one intermolecular and two intramolecular Na•••F short contacts are present. The 1H and 13C NMR spectra of the copper-ethene complexes show signals for the ethene ligands in the range of 4.84–4.96 ppm and 84.9–86.8 ppm respectively. The infrared spectra of the carbon monoxide complexes show CO stretching frequencies in the range of 2096–2120 cm–1. Both the NMR signals for the ethene ligands and infrared signals for the carbonyl ligands were found to show good correlations with the Hammett σp parameters of the substituents on the phenyl rings of the ligands. |
11/16/2015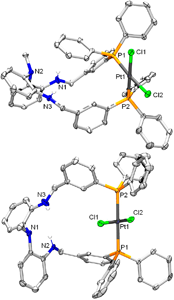 |
Congratulations to Andrea Pascui from the van der Vlugt's lab (Amsterdam, The Netherlands) for her recently published article in Eur. J. Inorg. Chem. (IF: 2.942) - linkMacrocyclic Platinum(II) Complexes with a Bifunctional Diphosphine LigandA. E. Pascui, K. van Rees, D. W. Zant, D. L. J. Broere, M. A. Siegler, J. I. van der Vlugt Abstract: The preparation and coordination chemistry of a ditopic ligand scaffold (1H2), containing both a phosphorus-donor (P) and a nitrogen-donor (N) binding pocket, is reported. The ligand was synthesized by reductive amination from 3-(diphenylphosphino)benzaldehyde and N-(2-aminophenyl)-N-methylbenzene-1,2-diamine. Selective coordination in the P-pocket was achieved for PtII, with careful control over the reaction conditions and precursor materials providing access to either the thermodynamic cis isomer, cis-PtCl2(1H2) (2), or the kinetic trans isomer, trans-PtCl2(1H2) (3). Both species have been fully characterized both in the solid state and in solution. Thermodynamic parameters for isomerization processes of 2 and 3 have been determined. Halide abstraction from 2 and 3 led to formation of cis-[Pt(CH3CN)(Cl)(1H2)]BF4 and trans-[Pt(CH3CN)(Cl)(1H2)]BF4. |
11/03/2015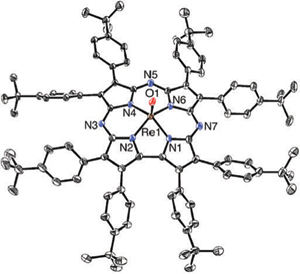 |
Congratulations to Jan Paulo Zaragoza from the Goldberg's lab for his recently published article in Chem. Commun. (IF: 6.834) - linkRhenium(V)–oxo corrolazines: isolating redox-active ligand reactivityJ. P. T. Zaragoza, M. A. Siegler, D. P. Goldberg Abstract: The synthesis of the first example of a third-row metallocorrolazine characterized by single crystal X-ray diffraction is reported. This ReV(O) porphyrinoid complex shows an exclusively ligand-based reactivity with strong acids and oxidizing agents. The one-electron oxidized π-radical-cation complex is capable of H-atom abstraction. |
10/29/2015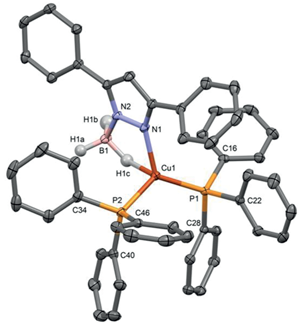 |
Congratulations to Thomas van Dijkman from the Bouwman lab (Leiden University, The Netherlands) for his recently published article in Eur. J. Inorg. Chem. (IF: 2.942) - linkBright Cyan Phosphorescence of a (Phosphane)copper(I) Complex of the Trihydridopyrazolylborate Ligand H3B(3,5-Ph2Pz)–T. F. van Dijkman, H. M. de Bruijn, M. A. Siegler, E. Bouwman Abstract: The ligand trihydrido(3,5-diphenylpyrazol-1-yl)borate([MpPh2]–) has been synthesized from 3,5-diphenylpyrazole and sodium borohydride in N,N-dimethylformamide. By using this ligand, three phosphane-stabilized copper(I) complexes [Cu(MpPh2)(L2)] [L2 = (PPh3)2, 1,2-bis(diphenylphosphanyl)ethane (dppe), or 1,2-bis(diphenylphosphanyl)benzene (dppbz)] were synthesized. The complexes were characterized by multinuclear NMR spectroscopy, IR spectroscopy, and X-ray crystallography. The crystal structures of these complexes show that the copper ions are in trigonal-pyramidal geometries with the apical position formed by agostic Cu–H interactions between the CuI center and one of the hydride atoms of the borate ligand. Complexes [Cu(MpPh2)(PPh3)2] and [Cu(MpPh2)(dppbz)] are mononuclear, whereas complex [Cu2(MpPh2)2(μ-dppe)2] is dinuclear with bridging dppe ligands. In the solid state, fluorescent emissions are observed in [Cu(MpPh2)(PPh3)2] and [Cu2(MpPh2)2(μ-dppe)2] but not in [Cu(MpPh2)(dppbz)], which exhibits bright cyan phosphorescence at room temperature that shifts to green when the sample is cooled to 77 K. The phosphorescence of [Cu(MpPh2)(dppbz)] is attributed to mixed interligand and metal-to-ligand charge transfer 3(MLCT + π–π*) excited states. |
09/25/2015 |
A new X-ray class will be tentatively offered for Spring 2016: A Theoretical and Experimental Approach to X-ray CrystallographyThe X-ray course will provide a comprehensive approach to X-ray structure determination (mostly concerned with small molecules) and its uses in Chemistry. The first segment of this course will cover all theoretical aspects of X-ray crystallography, i.e. crystals and crystallization, the nature of X-rays, the diffraction phenomenon of X-rays by crystals, symmetry and space groups, crystal structure analysis. Additionally, the course will provide laboratory experience for the students, involving hands-on instrumentation, experimental methodology to X-ray structure determination, structure solution/refinement, data analyses and publishing data. To whom it is aimed at?: Graduate students with a strong interest in Organic/Inorganic Chemistry, Materials Sciences, and Physics. Undergraduate students with a major in Chemistry are also encouraged. Semester: Tentatively Spring 2016 and subsequent years. Credit Hours: 3 Level: 600 Students: 15 (maximum) |
08/24/2015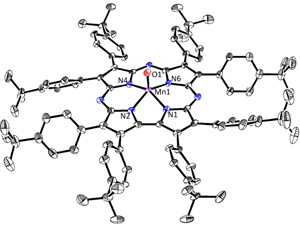 |
Congratulations to Regina Baglia from the Goldberg lab (JHU) for her recently published article in JACS (IF: 11.444) - linkMn(V)(O) versus Cr(V)(O) Porphyrinoid Complexes: Structural Characterization and Implications for Basicity Controlling H-Atom AbstractionR. A. Baglia, K. A. Prokop-Prigge, H. M. Neu, M. A. Siegler, D. P. Goldberg Abstract: Isomorphous crystals of MnV(O) and CrV(O) corrolazines were characterized by single crystal X-ray diffraction. Reactivity studies with H atom donors and separated PCET reagents show a dramatic difference in H atom abstracting abilities for these two complexes. The implied large difference in driving force is opposite the trend in redox potentials, indicating that basicity is a key factor in determining the striking difference in reactivity for two metal-oxo species in identical ligand environments. |
08/19/2015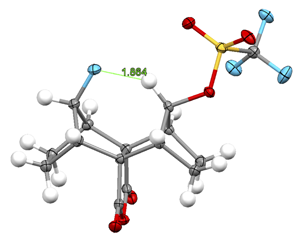 |
Congratulations to Mark Struble from the Lectka lab (JHU) for his recently published article in JACS (IF: 11.444) - linkSearch for a Symmetrical C-F-C Fluoronium Ion in Solution: Kinetic Isotope Effects, Synthetic Labeling, Computational, Solvent and Rate StudiesM. D. Struble, M. Gargiulo Holl, M. T. Scerba, M. A. Siegler, T. Lectka Abstract: Recently, we reported evidence for the generation of a symmetrical fluoronium ion (a [C—F—C]+ interaction) in solution from a cage-like precursor, relying primarily on a single isotopic-labeling experiment. Paraphrasing the axiom that a strong claim must be met by as much evidence as possible, we seek to expand upon our initial findings with comprehensive labeling studies, rate measurements, kinetic isotope effect (KIE) experiments, synthetic studies, and computations. We also chronicle the development of the system, our thought process and how it evolved from a tantalizing indication of fluoronium ion assistance in a dibromination reaction to the final, optimized system. Our experiments show secondary KIE experiments that are fully consistent with a transition state involving fluorine participation; this is also confirmed by a significant remote isotope effect. Paired with DFT calculations, the KIE experiments are indicative of the trapping of a symmetrical intermediate. Additionally, starting with an epimeric in-triflate precursor that hydrolyzes through a putative frontside SNi mechanism involving fluorine participation, KIE studies indicate that an identical intermediate is trapped (the fluoronium ion). Studies also show that the rate-determining step of the fluoronium forming SN1 reaction can be changed based on solvent and additives. We also report the synthesis of a non-fluorinated control substrate to measure a relative anchimeric role of the fluorine atom in hydrolysis versus μ-hydrido bridging. After extensive testing, we can make the remarkable conclusion that our system reacts solely through a “tunable” SN1 mechanism involving a fluoronium ion inter-mediate. Alternative scenarios, such as SN2 reactivity, do not occur even under forced conditions where they should be highly favored. |
05/15/2015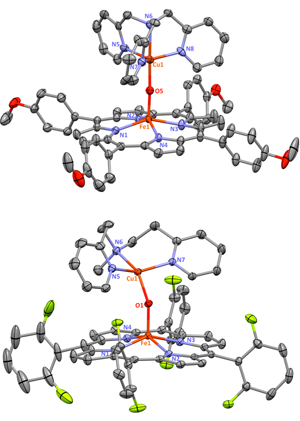 |
Congratulations to Shabnam Hematian from the Karlin lab (JHU) for her recently published article in JACS (IF: 11.444) - linkNitrogen Oxide Atom-Transfer Redox Chemistry; Mechanism of NO(g) to Nitrite Conversion Utilizing μ-oxo Heme-FeIII–O–CuII(L) ConstructsS. Hematian, I. Kenkel, T. E. Shubina, M. Dürr, J. J. Liu, M. A. Siegler, I. Ivanovic-Burmazovic, K. D. Karlin Abstract: While nitric oxide (NO, nitrogen monoxide) is a critically important signaling agent, its cellular concentrations must be tightly controlled, generally through its oxidative conversion to nitrite (NO2–) where it is held in reserve to be reconverted as needed. In part, this reaction is mediated by the binuclear heme a3/CuB active site of cytochrome c oxidase. In this report, the oxidation of NO(g) to nitrite is shown to occur efficiently in new synthetic μ-oxo heme-FeIII–O–CuII(L) constructs (L being a tridentate or tetradentate pyridyl/alkylamino ligand), and spectroscopic and kinetic investigations provide detailed mechanistic insights. Two new X-ray structures of μ-oxo complexes have been determined and compared to literature analogs. All μ-oxo complexes react with 2 mol equiv NO(g) to give 1:1 mixtures of discrete [(L)CuII(NO2–)]+ plus ferrous heme-nitrosyl compounds; when the first NO(g) equiv reduces the heme center and itself is oxidized to nitrite, the second equiv of NO(g) traps the ferrous heme thus formed. For one μ-oxo heme-FeIII–O–CuII(L) compound, the reaction with NO(g) reveals an intermediate species (“intermediate”), formally a bis-NO adduct, [(NO)(porphyrinate)FeII–(NO2–)–CuII(L)]+ (λmax = 433 nm), confirmed by cryo-spray ionization mass spectrometry and EPR spectroscopy, along with the observation that cooling a 1:1 mixture of [(L)CuII(NO2–)]+ and heme-FeII(NO) to −125 °C leads to association and generation of the key 433 nm UV–vis feature. Kinetic-thermodynamic parameters obtained from low-temperature stopped-flow measurements are in excellent agreement with DFT calculations carried out which describe the sequential addition of NO(g) to the μ-oxo complex. |
05/14/2015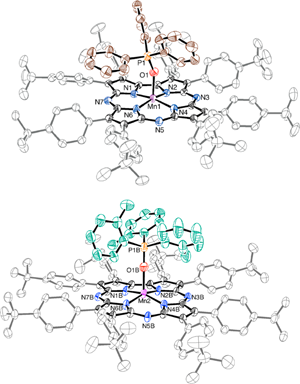 |
Congratulations to Jan Paulo Zaragoza from the Goldberg lab (JHU) for his recently published article in JACS (IF: 11.444) - linkStrong Inhibition of O-Atom Transfer Reactivity for MnIV(O)(π-Radical-Cation)(Lewis Acid) versus MnV(O) Porphyrinoid ComplexesJ. P. T. Zaragoza, R. A. Baglia, M. A. Siegler, D. P. Goldberg Abstract: The oxygen atom transfer (OAT) reactivity of two valence tautomers of a MnV(O) porphyrinoid complex was compared. The OAT kinetics of MnV(O)(TBP8Cz) (TBP8Cz = octakis(p-tert-butylphenyl)corrolazinato3–) reacting with a series of triarylphosphine (PAr3) substrates were monitored by stopped-flow UV–vis spectroscopy, and revealed second-order rate constants ranging from 16(1) to 1.43(6) × 104 M–1 s–1. Characterization of the OAT transition state analogues MnIII(OPPh3)(TBP8Cz) and MnIII(OP(o-tolyl)3)(TBP8Cz) was carried out by single-crystal X-ray diffraction (XRD). A valence tautomer of the closed-shell MnV(O)(TBP8Cz) can be stabilized by the addition of Lewis and Brønsted acids, resulting in the open-shell MnIV(O)(TBP8Cz•+):LA (LA = ZnII, B(C6F5)3, H+) complexes. These MnIV(O)(π-radical-cation) derivatives exhibit dramatically inhibited rates of OAT with the PAr3 substrates (k = 8.5(2) × 10–3 – 8.7 M–1 s–1), contrasting the previously observed rate increase of H-atom transfer (HAT) for MnIV(O)(TBP8Cz•+):LA with phenols. A Hammett analysis showed that the OAT reactivity for MnIV(O)(TBP8Cz•+):LA is influenced by the Lewis acid strength. Spectral redox titration of MnIV(O)(TBP8Cz•+):ZnII gives Ered = 0.69 V vs SCE, which is nearly +700 mV above its valence tautomer MnV(O)(TBP8Cz) (Ered = −0.05 V). These data suggest that the two-electron electrophilicity of the Mn(O) valence tautomers dominate OAT reactivity and do not follow the trend in one-electron redox potentials, which appear to dominate HAT reactivity. This study provides new fundamental insights regarding the relative OAT and HAT reactivity of valence tautomers such as MV(O)(porph) versus MIV(O)(porph•+) (M = Mn or Fe) found in heme enzymes. |
05/05/2015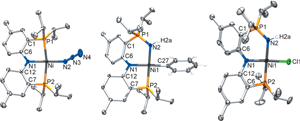 |
Congratulations to Vincent Vreeken from the van der Vlugt lab (University of Amsterdam, The Netherlands) for his recently published article in Angewandte Chemie International Edition (IF: 11.336) - linkC-H Activation of Benzene by a Photoactivated NiII(azide): Formation of a Transient Nickel Nitrido ComplexV. Vreeken, M. A. Siegler, B. de Bruin, J. N. H. Reek, M. Lutz, J. I. van der Vlugt Abstract: Photochemical activation of nickel-azido complex 2 [Ni(N3)(PNP)] (PNHP=2,2′-di(isopropylphosphino)-4,4′-ditolylamine) in neat benzene produces diamagnetic complex 3 [Ni(Ph)(PNPNH)], which is crystallographically characterized. DFT calculations support photoinitiated N2-loss of the azido complex to generate a rare, transient NiIV nitrido species, which bears significant nitridyl radical character. Subsequent trapping of this nitrido through insertion into the Ni-P bond generates a coordinatively unsaturated NiII imidophosphorane P=N donor. This species shows unprecedented reactivity toward 1,2-addition of a C-H bond of benzene to form 3. The structurally characterized chlorido complex 4 [Ni(Cl)(PNPNH)] is generated by reaction of 3 with HCl or by direct photolysis of 2 in chlorobenzene. This is the first report of aromatic C-H bond activation by a trapped transient nitrido species of a late transition metal. |
04/09/2015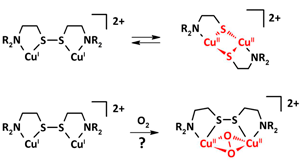 |
Congratulations to Dr. Ording-Wenker from the Bouwman lab (Leiden, The Netherlands) for her recently published article in Dalton Transactions (IF: 4.097) - linkCatalytic Catechol Oxidation by Copper Complexes: Development of a Structure-Activity RelationshipE.C.M. Ording-Wenker, M. A. Siegler, M. Lutz, E. Bouwman Abstract: A large library of CuII complexes with mononucleating and dinucleating ligands was synthesized to investigate their potential as catalysts for the catalytic oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC). X-ray structure determination of a number of these complexes revealed relatively large Cu…Cu distances and the formation of polymeric species. Comparison of the 3,5-DTBC oxidation rates showed that ligands that stabilize the biomimetic dinuclear CuII µ-thiolate complex also result in copper compounds that are much more active in the oxidation of 3,5-DTBC. This oxidation activity is however inhibited by the presence of chloride ions. The highest kcat that was observed was 6900 h–1, which is one of the highest turnover frequencies reported so far for catechol oxidation in CH3CN. |
04/06/2015 |
Congratulations to Heather Neu from the Goldberg lab (JHU) for her recently published article in JACS (IF: 11.444) - linkLight-Driven, Proton-Controlled, Catalytic Aerobic C–H Oxidation Mediated by a Mn(III) Porphyrinoid ComplexH. M. Neu, J. Jung, R. A. Baglia, M. A. Siegler, K. Ohkubo, S. Fukuzumi, D. P. Goldberg Abstract: The visible light-driven, catalytic aerobic oxidation of benzylic C–H bonds was mediated by a MnIII corrolazine complex. To achieve catalytic turnovers, a strict selective requirement for the addition of protons was established. The resting state of the catalyst was unambiguously characterized by X-ray diffraction as [MnIII(H2O)(TBP8Cz(H))]+, in which a single, remote site on the ligand is protonated. If two remote sites are protonated, however, reactivity with O2 is shut down. Spectroscopic methods revealed that the related MnV(O) complex is also protonated at the same remote site at −60°C, but undergoes valence tautomerization upon warming. |
04/02/2015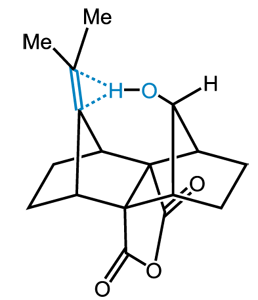 |
Congratulations to Mark Struble from the Lectka lab (JHU) for his recently published article in The Journal of Organic Chemistry (IF: 4.638 ) - linkSynthesis of a Tight Intramolecular OH---Olefin Interaction, Probed by IR, 1H NMR and Quantum ChemistryM. D. Struble, M. G. Holl, G. Coombs, M. A. Siegler, T. Lectka Abstract: We have synthesized a molecule containing a tight hydrogen bonding interaction between an alcohol and a non-conjugated pi-system. The strength of this hydrogen bond results in a large red shift, nearly 189 cm-1, on the alcohol stretching frequency in the IR spectrum in comparison to a free alcohol control. The interaction is notable in that it possesses a better defined intramolecular hydrogen bond compared to the usual molecules for which it is noted, such as syn-7-norbornenol. This interaction was well studied through the use of IR and NMR spectroscopy, X-ray crystallography and molecular modeling calculations. |
03/25/2015 |
Congratulations to Pankaj Kumar from the Nam group (Department of Chemistry and Nano Science, Ewha Womans University, South Korea) for his recently published article in JACS (IF: 11.444) - linkReactions of Co(III)–Nitrosyl Complexes with Superoxide and Their Mechanistic InsightsP. Kumar, Y-M. Lee, Y J. Park, M. A. Siegler, K. D. Karlin, W. Nam Abstract: New CoIII-nitrosyl complexes bearing N-tetramethylated cyclam (TMC) ligands, [(12-TMC)CoIII(NO)]2+ (1) and [(13-TMC)CoIII(NO)]2+ (2), were synthesized via [(TMC)CoII(CH3CN)]2+ plus NO(g) reactions. Spectroscopic and structural characterization shows that these compounds bind the nitrosyl moiety in a bent end-on fashion. The CoIII-nitrosyl complexes, 1 and 2, reacted with KO2/2.2.2-Cryptand and produced [(12-TMC)CoII(NO2)]+ (3) and [(13-TMC)CoII(NO2)]+ (4), respectively; these possess O,O’-chelated nitrito ligands. Mechanistic studies using 18O-labeled superoxide (18O2•–) demonstrate that one oxygen atom in the nitrito ligand derives from superoxide and dioxygen produced comes from the other superoxide oxygen atom. Evidence supporting the formation of a Co-peroxynitrite intermediate is also presented. |
03/23/2015 |
Congratulations to Danny Broere from the van der Vlugt group (University of Amsterdam, The Netherlands) for his recently published article in Chemistry - A European Journal (IF: 5.696) - linkDinuclear Palladium Complexes with Two Ligand-CenteredRadicals and a Single Bridging Ligand: Subtle Tuning of MagneticPropertiesD. L. J. Broere, S. Demeshko, B. de Bruin, E. A. Pidko, J. N. H. Reek, M. A. Siegler, M. Lutz, J. I. van der Vlugt Abstract: The facile and tunable preparation of unique dinuclear [(L⋅)Pd-X-Pd(L⋅)] complexes (X=Cl or N3), bearing a ligand radical on each Pd, is disclosed, as well as their magnetochemistry in solution and solid state is reported. Chloride abstraction from [PdCl(NNOISQ)] (NNOISQ=iminosemiquinonato) with TlPF6 results in an unusual monochlorido-bridged dinuclear open-shell diradical species, [{Pd(NNOISQ)}2(μ-Cl)]+, with an unusually small Pd-Cl-Pd angle (ca. 93°, determined by X-ray). This suggests an intramolecular d8–d8 interaction, which is supported by DFT calculations. SQUID measurements indicate moderate antiferromagnetic spin exchange between the two ligand radicals and an overall singlet ground state in the solid state. VT EPR spectroscopy shows a transient signal corresponding to a triplet state between 20 and 60 K. Complex 2 reacts with PPh3 to generate [Pd(NNOISQ)(PPh3)]+ and one equivalent of [PdCl(NNOISQ)]. Reacting an 1:1 mixture of [PdCl(NNOISQ)] and [Pd(N3)(NNOISQ)] furnishes the 1,1-azido-bridged dinuclear diradical [{Pd(NNOISQ)}2(κ1-N;μ-N3]+, with a Pd-N-Pd angle close to 127° (X-ray). Magnetic and EPR measurements indicate two independent S=equation image spin carriers and no magnetic interaction in the solid state. The two diradical species both show no spin exchange in solution, likely because of unhindered rotation around the Pd-X-Pd core. This work demonstrates that a single bridging atom can induce subtle and tunable changes in structural and magnetic properties of novel dinuclear Pd complexes featuring two ligand-based radicals. |
03/16/2015 |
Congratulations to Dr. Guo from the Katz lab (JHU) for his recently published article in Angewandte Chemie International Edition (IF: 11.336) - linkVisible-Light-Triggered Molecular Photoswitch Based on Reversible E/Z Isomerization of a 1,2-Dicyanoethene DerivativeX. Guo, J. Zhou, M. A. Siegler, A. E. Bragg, H. E. Katz Abstract: A designed bis(dithienyl) dicyanoethene-based, strictly E/Z photoswitch (4TCE) operates through state-selective (E and Z isomer) photoactivation with visible light. The E and Z isomers of 4TCE exhibit remarkably different spectroscopic characteristics, including a large separation (70 nm) in their absorption maxima (λmax) and a 2.5-fold increase in molar extinction coefficient from cis to trans. The energetically stable trans form can be completely converted to the cis form within minutes when exposed to white light, whereas the reverse isomerization occurs readily upon irradiation by blue light (λ<480 nm) or completely by thermal conversion at elevated temperatures. These features together with excellent thermal stability and photostability of both isomers make this new E/Z photoswitch a promising building block for photoswitchable materials that operate without the need for UV light. |
03/12/2015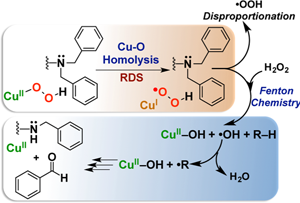 |
Congratulations to Dr. Kim from the Karlin lab (JHU) for her recently published article in JACS (IF: 11.444) - linkAmine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton ChemistryS. Kim, J. W. Ginsbach, J. Yoon Lee, R. L. Peterson, J. J. Liu, M. A. Siegler, A. A. Sarjeant, E. I. Solomon, K. D. Karlin Abstract: Copper(II) hydroperoxide species are significant intermediates in processes such as fuel cells and (bio)chemical oxidations, all involving stepwise reduction of molecular oxygen. We previously reported a CuII-OOH species that performs oxidative N-dealkylation on a dibenzylamino group that is appended to the 6-position of a pyridyl donor of a tripodal tetradentate ligand. To obtain insights into the mechanism of this process, reaction kinetics and products were determined employing ligand substrates with various para-substituent dibenzyl pairs (-H,-H; -H,-Cl; -H,-OMe, and -Cl,-OMe), or with partially or fully deuterated dibenzyl N-(CH2Ph)2 moieties. A series of ligand–copper(II) bis-perchlorate complexes were synthesized, characterized, and the X-ray structures of the -H,-OMe analogue were determined. The corresponding metastable CuII-OOH species were generated by addition of H2O2/base in acetone at −90 °C. These convert (t1/2 ≈ 53 s) to oxidatively N-dealkylated products, producing para-substituted benzaldehydes. Based on the experimental observations and supporting DFT calculations, a reaction mechanism involving dibenzylamine H-atom abstraction or electron-transfer oxidation by the CuII-OOH entity could be ruled out. It is concluded that the chemistry proceeds by rate limiting Cu-O homolytic cleavage of the CuII-(OOH) species, followed by site-specific copper Fenton chemistry. As a process of broad interest in copper as well as iron oxidative (bio)chemistries, a detailed computational analysis was performed, indicating that a CuIOOH species undergoes O–O homolytic cleavage to yield a hydroxyl radical and CuIIOH rather than heterolytic cleavage to yield water and a CuII-O•– species. |
03/11/2015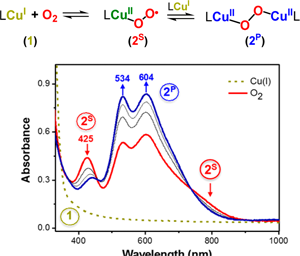 |
Congratulations to Dr. Kim from the Karlin lab (JHU) for her recently published article in JACS (IF: 11.444) - linkA N3S(thioether)-Ligated CuII-Superoxo with Enhanced ReactivityS. Kim, J. Yoon Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon, K. D. Karlin Abstract: Previous efforts to synthesize a cupric superoxide complex possessing a thioether donor have resulted in the formation of an end-on trans-peroxo-dicopper(II) species, [{(Ligand)CuII}2(μ-1,2-O22–)]2+. Redesign/modification of previous N3S tetradentate ligands has now allowed for the stabilization of the monomeric, superoxide product possessing a S(thioether) ligation, [(DMAN3S)CuII(O2•–)]+ (2S), as characterized by UV–vis and resonance Raman spectroscopies. This complex mimics the putative CuII(O2•–) active species of the copper monooxygenase PHM and exhibits enhanced reactivity toward both O–H and C–H substrates in comparison to close analogues [(L)CuII(O2•–)]+, where L contains only nitrogen donor atoms. Also, comparisons of [(L)CuII/I]n+ compound reduction potentials (L = various N4 vs DMAN3S ligands) provide evidence that DMAN3S is a weaker donor to copper ion than is found for any N4 ligand-complex. |
03/09/2015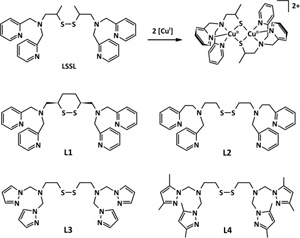 |
Congratulations to Dr. Ording-Wenker from the Bouwman lab (Leiden, The Netherlands) for her recently published article in Inorganica Chimica Acta (IF: 2.041) - linkCoordination of new disulfide ligands to CuI and CuII: Does a CuIIμ-thiolate complex form?E. C.M. Ording-Wenker, M. A. Siegler, E. Bouwman Abstract: Interest in dinuclear CuIIμ-thiolate and CuI disulfide complexes is triggered by their similarity with the CuA site and the possibility to control this redox equilibrium. Three new disulfide ligands L1, L3 and L4 were synthesized and reacted with CuI salts to investigate whether thiolate or disulfide species would form. The nature of L1 precludes the formation of CuIIμ-thiolate species, resulting in the formation of [CuI2(L1)(CH3CN)](BF4)2 which was characterized via single crystal X-ray crystallography. Pyrazole-containing ligands L3 and L4 form CuI complexes that are stable in solution in air for hours with half-wave potentials of approximately +0.55 V versus Ag/AgCl, indicating high stability of the CuI state rather than the CuII state. The half-wave potentials of the CuI complexes with L1 and L2 are less positive, indicating that in order to allow formation of both CuIIμ-thiolate and CuI disulfide species, a half-wave potential of roughly 0 V versus Ag/AgCl would be ideal. Furthermore, CuII crystal structures with L1, L2, L3 and L4 and different counterions were compared and analyzed. Pyrazolyl-containing ligands L3 and L4 form complexes that are very similar to the complexes with pyridyl-containing ligands L1 and L2. |
02/11/2015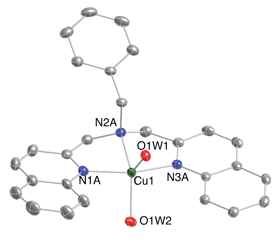 |
Congratulations to Dr. Rolle from the Karlin lab (JHU) for his recently published article in JACS (IF: 11.444) - linkLewis Acid-Induced Change from Four- to Two-Electron Reduction of Dioxygen Catalyzed by Copper Complexes Using Scandium TriflateS. Kakuda, C. Rolle, K. Ohkubo, M. A. Siegler, K. D. Karlin , S. Fukuzumi Abstract: Mononuclear copper complexes, [(tmpa)CuII(CH3CN)](ClO4)2 (1, tmpa = tris(2-pyridylmethyl)amine) and [(BzQ)CuII(H2O)2](ClO4)2 (2, BzQ = bis(2-quinolinylmethyl)benzylamine)], act as efficient catalysts for the selective two-electron reduction of O2 by ferrocene derivatives in the presence of scandium triflate (Sc(OTf)3), in acetone, whereas 1 catalyzes the four-electron reduction of O2 by the same reductant in the presence of Brønsted acids such as triflic acid. Following formation of the peroxo-bridged dicopper(II) complex [(tmpa)CuII(O2)CuII(tmpa)]2+, the two-electron reduced product of O2 with Sc3+ is observed to be scandium peroxide ([Sc3+(O22–)]+). In the presence of three equiv of hexamethylphosphoric triamide (HMPA), [Sc3+(O22–)]+ was oxidized by [Fe(bpy)3]3+ (bpy = 2,2’-bipyridine) to the known superoxide species [(HMPA)3Sc3+(O2•–)]2+ as detected by EPR spectroscopy. A kinetic study revealed that the ratedetermining step of the catalytic cycle for the two-electron reduction of O2 with 1 is electron transfer from Fc* to 1 to give a cuprous complex which is highly reactive toward O2, whereas the rate-determining step with 2 is changed to the reaction of the cuprous complex with O2 following electron transfer from ferrocene derivatives to 2. The explanation for the change in catalytic O2-reaction stoichiometry from four electron with Brønsted acids to two-electron reduction in the presence of Sc3+ and also for the change in the rate-determining step is clarified based on a kinetics interrogation of the overall catalytic cycle as well as each step of the catalytic cycle with study of the observed effects of Sc3+ on copper-oxygen intermediates. |
01/06/2015 |
Congratulations to Dr. Surampudi from the Klausen lab (JHU) for his recently published article in Chemical Science (IF: 8.601) - linkIncreased Carrier Mobility in End-Functionalized OligosilanesS. Surampudi, M.-L. Yeh, M. A. Siegler, J. F. Martinez Hardigree, T. A. Kasl, H. E. Katz, R.S. Klausen Abstract: We show that a class of oligosilane-arene σ,π-hybrid materials exhibits distinct and enhanced solid-state electronic properties relative to its parent components. In the single crystal structure, the σ-conjugation axis of one molecule points towards the π-face of a neighboring molecule due to an unusual gauche conformation. This organization is hypothesized to be beneficial for charge transport. We show that solution-deposited crystalline films of the hybrid materials show up to a 100-fold increase in space-charge limited current (SCLC) mobility relative to literature reports of photoinduced hole transport in oligosilane films. The discovery that σ,π-hybrids are more than the sum of their parts offers a design opportunity for new materials. |
01/05/2015 |
Happy New Year! and let 2015 be an even more fruitful year than the previous one. 2014 has been a good year as our X-ray facility lab has been involved in 22 publications, including 1 Nature Communications, 1 Angewandte Chemie International Edition, and 5 JACS. The full list of publications for 2014 is available here: link |
12/18/2014 |
Congratulations to Dr. Seungwoo Hong from the Wonwoo Nam group (Department of Chemistry, Division of Nano Sciences and Centre for Biomimetic Systems, Ewha Womans University, Seoul, South Korea) for his recently published article in Nature Communications (IF: 10.742) - linkCrystallographic and spectroscopic characterization and reactivities of a mononuclear non-haem iron(III)-superoxo complexS. Hong, K. D. Sutherlin, J. Park, E. Kwon, M. A. Siegler, E. I. Solomon, W. Nam Abstract: Mononuclear non-haem iron(III)-superoxo species (FeIII-O2−·) have been implicated as key intermediates in the catalytic cycles of dioxygen activation by non-haem iron enzymes. Although non-haem iron(III)-superoxo species have been trapped and characterized spectroscopically in enzymatic and biomimetic reactions, no structural information has yet been obtained. Here we report the isolation, spectroscopic characterization and crystal structure of a mononuclear side-on (η2) iron(III)-superoxo complex with a tetraamido macrocyclic ligand. The non-haem iron(III)-superoxo species undergoes both electrophilic and nucleophilic oxidation reactions, as well as O2-transfer between metal complexes. In the O2-transfer reaction, the iron(III)-superoxo complex transfers the bound O2 unit to a manganese(III) analogue, resulting in the formation of a manganese(IV)-peroxo complex, which is characterized structurally and spectroscopically as a mononuclear side-on (η2) manganese(IV)-peroxo complex. The difference in the redox distribution between the metal ions and O2 in iron(III)-superoxo and manganese(IV)-peroxo complexes is rationalized using density functional theory calculations. |
12/02/2014 |
Congratulations to Dr. Fry from the McQueen group for her recently published article in Journal of Solid State Chemistry (IF: 2.200) - linkUnique edge-sharing sulfate-transition metal coordination in Na2M(SO4)2 (M=Ni and Co)A. M. Fry, O. T. Sweeney, W. A. Phelan, N. Drichko, M A. Siegler, T. M. McQueen Abstract: Two new compounds, Na2Ni(SO4)2 and Na2Co(SO4)2, were synthesized and their structure and properties were characterized. They form a new structure type, containing a bidentate coordination of sulfate to the transition metal center, which was determined via single crystal X-ray diffraction combined with model refinements to both laboratory X-ray and time-of-flight neutron powder diffraction data. The compounds were both found to crystallize in the C2/c space group with Z=24 and a unit cell of a=23.3461(3) Å, b=10.3004(1) Å, c=17.4115(2) Å, β=98.8659(9)°, and V=4136.99(8) Å3 for the cobalt analog and a=23.2253(1) Å, b=10.26155(6) Å, c=17.3353(1) Å, β=99.0376(5)°, and V=4080.20(5) Å3 for the nickel analog. Magnetization measurements show that the transition metal centers have negligible interactions with neighboring sites. Infrared and Raman spectroscopies were used to further probe the unique sulfate-transition metal coordination, and confirm the bidentate binding motif. The resulting pseudo-trigonal bipyramidal coordination produces vivid violet, Na2Co(SO4)2, and yellow, Na2Ni(SO4)2, colors that were probed by diffuse reflectance. |
12/01/2014 |
Congratulations to Danny Broere from the van der Vlugt group (University of Amsterdam, The Netherlands) for his recently published article in Angewandte Chemie International Edition (IF: 13.734) - linkRedox-Active Ligand-Induced Homolytic Bond ActivationD. L. J. Broere, L. L. Metz, B. de Bruin, J. N. H. Reek, M. A. Siegler, J. I. van der Vlugt Abstract: Coordination of the novel redox-active phosphine-appended aminophenol pincer ligand (PNOH2) to PdII generates a paramagnetic complex with a persistent ligand-centered radical. The complex undergoes fully reversible single-electron oxidation and reduction. Homolytic bond activation of diphenyldisulfide by the single-electron reduced species leads to a ligand-based mixed-valent dinuclear palladium complex with a single bridging thiolate ligand. Mechanistic investigations support an unprecedented intramolecular ligand-to-disulfide single-electron transfer process to induce homolytic S-S cleavage, thereby releasing a thiyl (sulfanyl) radical. This could be a new strategy for small-molecule bond activation. |
11/24/2014 |
Congratulations to Ashot Khrimian et al. from USDA (Maryland, USA) for their recently published article in J. Chem. Ecol. (IF: 2.239) - linkDetermination of the Stereochemistry of the Aggregation Pheromone of Harlequin Bug, Murgantia histrionicaA. Khrimian, S. Shirali, K. E. Vermillion, M. A. Siegler, F. Guzman, K. Chauhan, J. R. Aldrich, D. C. Weber Abstract: Preparation of a complete stereoisomeric library of 1,10-bisaboladien-3-ols and selected 10,11-epoxy-1-bisabolen-3-ols was pivotal for the identification of the aggregation pheromone of the brown marmorated stink bug, Halyomorpha halys. Herein, we describe syntheses of the remaining 10,11-epoxy-1-bisabolen-3-ols, and provide additional evidence on the assignment of relative and absolute configurations of these compounds by single-crystal X-ray crystallography of an intermediate, (3S,6R,7R,10S)-1-bisabolen-3,10,11-triol. To demonstrate the utility of this stereoisomeric library, we revisited the aggregation pheromone of the harlequin bug, Murgantia histrionica, and showed that the male-produced pheromone consists of two stereoisomers of 10,11-epoxy-1-bisabolen-3-ol. Employment of eight cis-10,11-epoxy-1-bisabolen-3-ol stereoisomeric standards, two enantioselective GC columns, and NMR spectroscopy enabled the identification of these compounds as (3S,6S,7R,10S)-10,11-epoxy-1-bisabolen-3-ol and (3S,6S,7R,10R)-10,11-epoxy-1-bisabolen-3-ol, which are produced by M. histrionica males in 1.4:1 ratio. |
11/24/2014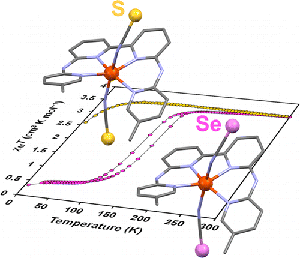 |
Congratulations to Sipeng Zheng from the Bonnet group (Leiden, The Netherlands) for his recently published article in Inorg. Chem. (IF: 4.794) - linkInfluence of Selenocyanate Ligands on the Transition Temperature and Cooperativity of bapbpy-Based Fe(II) Spin-Crossover CompoundsS. Zheng, M. A. Siegler, O. Roubeau, S. Bonnet Synopsis: Eight new molecular iron(II) compounds [Fe(R2bapbpy)(NCSe)2] are described, five of which show spin-crossover properties. The selenocyanate axial ligands increase the transition temperatures in these complexes, compared to the previously described thiocyanate analogues [Fe(R2bapbpy)(NCS)2]. One of the five bis(selenocyanato) SCO compounds is more cooperative than its thiocyanato analogue. The influence of sulfur replacement by selenium on the cooperativity of the spin crossover is discussed. |
10/23/2014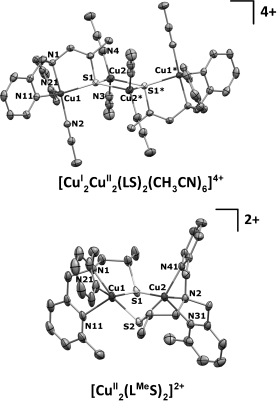 Displacement ellipsoid plots (50 % probability level) of the cationic parts of [CuI2CuII2(LS)2(CH3CN)6](BF4)4 and [CuII2(LMeS)2](BF4)2 at 100(2) and 110(2) K, respectively. All hydrogen atoms are omitted for clarity. Symmetry operation *=1−x, 1−y, 2−z. |
Congratulations to Erica Ording-Wenker from the Bouwman group (Leiden, The Netherlands) for her recently published article in Chemistry: A European Journal (IF: 5.696) - linkProtonation of a Biologically Relevant CuII μ-Thiolate Complex: Ligand Dissociation or Formation of a Protonated CuI Disulfide Species?E. C. M. Ording-Wenker, M. van der Plas, M. A. Siegler, C. Fonseca Guerra, E. Bouwman Abstract: The proton-induced electron-transfer reaction of a CuII μ-thiolate complex to a CuI-containing species has been investigated, both experimentally and computationally. The CuII μ-thiolate complex [CuII2(LMeS)2]2+ is isolated with the new pyridyl-containing ligand LMeSSLMe, which can form both CuII thiolate and CuI disulfide complexes, depending on the solvent. Both the CuII and the CuI complexes show reactivity upon addition of protons. The multivalent tetranuclear complex [CuI2CuII2(LS) 2(CH3CN)6]4+ crystallizes after addition of two equivalents of strong acid to a solution containing the μ-thiolate complex [CuII2(LS)2]2+ and is further analyzed in solution. This study shows that, upon addition of protons to the CuII thiolate compound, the ligand dissociates from the copper centers, in contrast to an earlier report describing redox isomerization to a CuI disulfide species that is protonated at the pyridyl moieties. Computational studies of the protonated CuII μ-thiolate and CuI disulfide species with LSSL show that already upon addition of two equivalents of protons, ligand dissociation forming [CuI(CH3CN)4]+ and protonated ligand is energetically favored over conversion to a protonated CuI disulfide complex. |
10/16/2014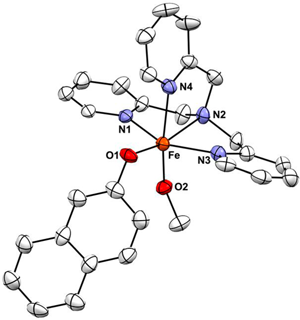 Displacement ellipsoid plot (50% probability level) of one of the two crystallographically independent [Fe(TPA)(2-naphtholate)-(OCH3)]+ cations at 110(2) K. The H atoms were omitted for the sake of clarity. |
Congratulations to Prof. Will Kerber (Bucknell University) for his recently published article in Inorg. Chem. (IF: 4.794) - linkSpeciation of Ferric Phenoxide Intermediates during the Reduction of Iron(III)−μ-Oxo Dimers by HydroquinoneW. D. Kerber, K. A. Perez, C. Ren, M. A. Siegler Abstract: Iron(III) is chelated by tris(pyridylmethyl)amine to form [Fe(TPA)] |
10/14/2014 |
THE WEBSITE HAS A NEW DESIGN! It has been tested on Google Chrome, Mozilla Firefox and Microsoft Internet Explorer. |
10/13/2014 |
IT’S ALL ABOUT ORIENTATION: congratulations to Sahu et al. (Goldberg group) for the article "Direct Observation of a Nonheme Iron(IV)–Oxo Complex That Mediates Aromatic C–F Hydroxylation" hitting the JACS Spotlights - link |
09/25/2014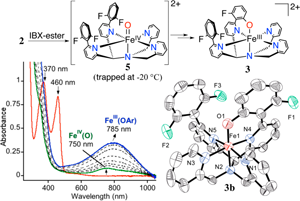 Overall scheme for aromatic hydroxylation (top), UV−vis spectral changes for 2a + IBX-ester over 50 min in CH3CN at 23 °C (bottom left), displacement ellipsoid plot (50% probability) for the dication of 3b (bottom right). H-atoms were omitted for clarity |
Congratulations to Sumit Sahu from the Goldberg group for his recently published article in JACS (IF: 10.677) - linkDirect Observation of a Nonheme Iron(IV)–Oxo Complex That Mediates Aromatic C–F HydroxylationS. Sahu, M. G. Quesne, C. G. Davies, M. Dürr, I. Ivanović-Burmazović, M. A. Siegler, G. N. L. Jameson, S. P. de Visser, D. P. Goldberg Abstract: The synthesis of a pentadentate ligand with strategically designed fluorinated arene groups in the second coordination sphere of a nonheme iron center is reported. The oxidatively resistant fluorine substituents allow for the trapping and characterization of an FeIV(O) complex at −20 °C. Upon warming of the FeIV(O) complex, an unprecedented arene C–F hydroxylation reaction occurs. Computational studies support the finding that substrate orientation is a critical factor in the observed reactivity. This work not only gives rare direct evidence for the participation of an FeIV(O) species in arene hydroxylation but also provides the first example of a high-valent iron–oxo complex that mediates aromatic C–F hydroxylation. |
08/08/2014 |
The Service page has been updated. It now includes a more refined list of services, a description of the four different available packages (numbered I, II, III and IV), and a pricing scheme for JHU and non-JHU users (including academic and non-academic). |
08/07/2014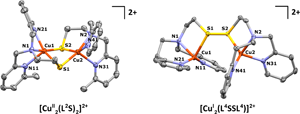 Displacement ellipsoid plots (50% probability level) of the cationic parts of [CuII2(L2S)2](BF4)2·C3H6O and [CuI2(L4SSL4)](BF4)2 at 110(2) K. Hydrogen atoms are omitted for clarity. |
Congratulations to Erica Ording-Wenker from the Bouwman group (Leiden, The Netherlands) for her recently published article in Inorganic Chemistry (IF: 4.794) - linkThermodynamics of the CuII μ-Thiolate and CuI Disulfide Equilibrium: A Combined Experimental and Theoretical StudyE. C.M. Ording-Wenker, M. van der Plas, M. A. Siegler, S. Bonnet, F. M. Bickelhaupt, C. Fonseca Guerra, E. Bouwman Abstract: The redox equilibrium between dinuclear CuII μ-thiolate and CuI disulfide structures has been analyzed experimentally and via DFT calculations. Two new ligands, L2SSL2 and L4SSL4, and their CuII μ-thiolate and CuI disulfide complexes were synthesized. For L2SSL2, these two redox-isomeric copper species are shown to be in equilibrium, which depends on both temperature and solvent. For L4SSL4 the μ-thiolate species forms as the kinetic product and further evolves into the disulfide complex under thermodynamic control, which creates the unprecedented possibility to compare both species under the same reaction conditions. The energies of the μ-thiolate and disulfide complexes for two series of related ligands have been calculated with DFT; the results rationalize the experimentally observed structures, and emphasize the important role that steric requirements play in the formation of the CuII thiolate structure. |
07/07/2014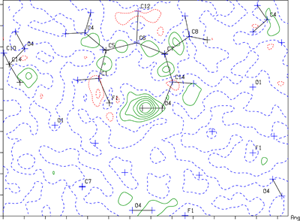 Contoured difference Fourier map drawn in the plane defined by O4 H4 F1. The location of the H atom is indicated by the contoured green circles near O4. |
Congratulations to Mark Struble from the Lectka group (JHU) for his recently published article in Angewandte Chemie International Edition (IF: 13.734) - linkSearch for a Strong, Virtually “No-Shift” Hydrogen Bond: A Cage Molecule with an Exceptional OH...F InteractionM. D. Struble, C. Kelly, M. A. Siegler, T. Lectka Abstract: Reported herein is the synthesis of a molecule containing an unusually strong hydrogen bond between an OH donor and a covalent F acceptor, a heretofore somewhat ill-defined if not controversial interaction. This unique hydrogen bond is to a large extent a product of the tight framework of the rigid caged system. Remarkably, the interaction shows little to no perceptible shift in the OH stretch of the IR spectrum relative to appropriate nonhydrogen-bound standards in fairly non-interactive solvents. This fascinating example of what has been termed a virtual “no-shift” hydrogen bond is investigated through NMR (coupling constants, isotopic chemical shift perturbations, proton exchange rates) and IR studies which all tell a consistent story. |
07/07/2014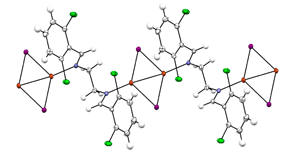 Displacement ellipsoid plot (50% probability level) of 2CuI.bis(imine) complex at 110(2) K. |
Congratulations to Cody Pitts from the Lectka group (JHU) for his recently published article in JACS (IF: 10.677) - linkDirect, Catalytic Monofluorination of sp3 C–H Bonds: A Radical-Based Mechanism with Ionic SelectivityC. R. Pitts, S. Bloom, R. Woltornist, D. Auvenshine, L. R. Ryzhkov, M. A. Siegler, T. Lectka Abstract: Recently, our group unveiled a system in which an unusual interplay between copper(I) and Selectfluor effects mild, catalytic sp3 C-H fluorination. Herein, we report a detailed reaction mechanism based on exhaustive EPR, 19F NMR, UV-vis, electrochemical, kinetic, synthetic, and computational studies that, to our surprise, was revealed to be a radical chain mechanism in which copper acts as an initiator. Furthermore, we offer an explanation for the notable but curious preference for monofluorination by ascribing an ionic character to the transition state |
Copyright © 2009-2014 - JHU X-ray Crystallography Facility v2.0 - Maxime Siegler